Autora: Luciana Nascimento Garcia
Colaboradores: Karoline Prado, Larissa Andrade Correa da Silva e Paulo Roberto de Pauli
O que é fenilcetonúria?
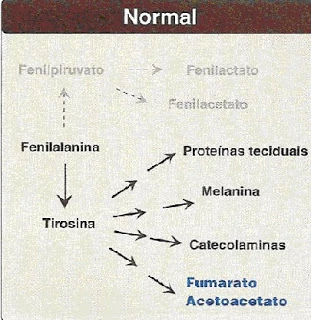
É uma doença hereditária de caráter autossômico recessivo, ou seja, que passa de pai para filho numa taxa de 25% com uma proporção global média de um caso para cada dez mil nascimentos. Caracteriza-se pela falta ou diminuição de uma enzima presente no fígado, a fenilalanina-hidroxilase (FAH), que é responsável por transformar um aminoácido essencial do nosso organismo, chamado fenilalanina (FAL) em tirosina (tyr). Como consequência, a fenilalanina se acumulará no sangue e tecido podendo, muitas vezes, acarretar em sequelas graves a nível cerebral.
A toxicidade da fenilalanina
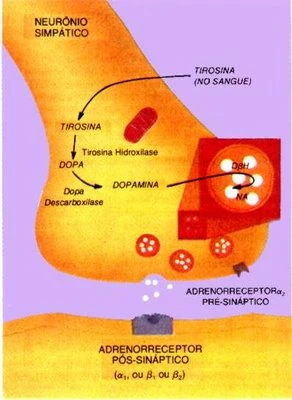
A fenilalanina está presente nos alimentos proteicos e, após a ingestão, ela possui dois destinos: o primeiro é ser depositado como aminoácido nos tecidos e no sangue e o segundo, ser transformado no aminoácido tirosina. Nas pessoas que possuem essa doença, o segundo destino não ocorre, e o excesso de fenilalanina é convertido em ácido pirúvico, presente na urina e no suor. Esse ácido inibirá a produção de certas gorduras, afetando assim, o crescimento cerebral, a formação de bainha de mielina nos neurônios e de neurotransmissores, uma vez que, o ácido pirúvico impede a entrada dos aminoácidos que são importantes na síntese de hormônios como serotonina e catecolaminas (adrenalina, noradrenalina e dopamina).
{kind=link}
{kind=link}
{kind=link}
Descrição: Liberação de neurotransmissores pelo neurônio pré simpático. Fonte:http://pkubiobio.blogspot.com.br/
Diagnóstico
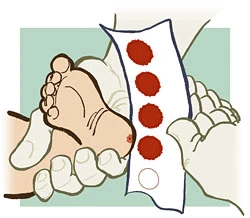
O diagnóstico pode e deve ser realizado no recém-nascido através do Teste do Pezinho, na própria maternidade. Este teste é gratuito e é um direito de todo cidadão. Ele consiste numa triagem neonatal na qual é coletada gotas de sangue do calcanhar do bebê.
{kind=link}
Descrição: Coleta de sangue do calcanhar do bebê para realização do Teste do Pezinho. Fonte:http: //blogbionut.blogspot.com.br/
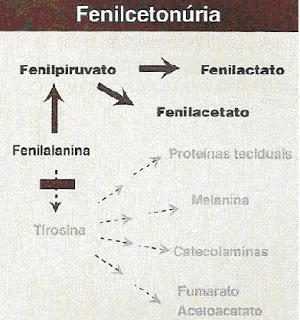
O resultado positivo acontece quando os níveis de fenilalanina são maiores que 2mmg/dl e há confirmação por uma segunda análise das quantidades de fenilalanina e tirosina sendo a razão dos dois igual a 3. Nos fenilcetonúricos, a tyr está com níveis diminuídos e há excreção elevada de metabólitos como o fenilpiruvato, fenilactato e fenilacetato.
Quando não for realizado o teste do pezinho, a fenilcetonúria pode ser diagnosticada por meio de análise laboratorial relacionada aos sintomas da doença, que podem aparecer aproximadamente aos 4 ou 6 meses de vida.
As pessoas que possuem essa doença devem fazer acompanhamento dos níveis de fenilalanina da seguinta forma:
- Quinzenalmente, se forem menores de 1 ano de idade;
- Mensalmente, no caso das grávidas;
- Período indeterminado, nos demais.
Sintomas
Uma das funções da tirosina no organismo é de formar a melanina (responsável pela nossa pigmentação) quando sofre hidroxilação (acrescenta OH no seu composto) pela enzima tirosinase. Como um indivíduo com fenilcetonúria não consegue formar tirosina, ele possui cabelos e pele claros. Inflamações na pele, complicações neurológicas e, mais raramente, autismo também são sintomas consequentes da fenilcetonúria.
Os metabólitos fenillactato, fenilacetato e fenilpiruvato, não são perceptíveis na urina das pessoas sem essa doença, porém, o aumento significativo desses metabólitos num fenilcetonúrico resultam em graves consequências ao sistema nervoso, sendo possível perceber devido a isso distúrbios cognitivos como déficit de atenção, convulsões e, principalmente, retardo mental. Ainda podem aparecer sintomas como odor corpóreo característico, hiperatividade, tremor, microcefalia (cabeça menor do que a do que o tamanho típico para a idade do feto ou criança) e crescimento comprometido do corpo.
Tratamento
Algo muito relevante e característico dessa doença é que é uma das poucas doenças de caráter genético na qual, atuando preventivamente através do diagnóstico e tratamento, pode-se evitar o retardo mental. Faz-se necessário ter uma dieta alimentar adequada por toda a vida, sendo que o não cumprimento disso pode acarretar em danos ao portador da doença.
As dietas dos bebês fenilcetonúricos são baseadas em uma mescla de aminoácidos acrescidos de carboidratos, gorduras, vitaminas e minerais inseridas no começo do tratamento e usadas paralelas ao leite materno. O bebê deve ter acompanhamento médico frequente e os pais devem ser orientados sobre a necessidade de seguir tais recomendações.
Outra alternativa de dieta para os portadores de fenilcetonúria são os hidrolisados proteicos com baixo teor de fenilalanina. Eles podem ser usados em qualquer faixa etária e tem como vantagens, em relação à mescla de aminoácidos, serem quase que inodoros e permitirem o acréscimo de sabor e aroma a esse composto.
Os alimentos que os portadores de fenilcetonúria podem ingerir são aqueles com níveis baixos de FEL. Alguns deles são: certos doces (mel, balas de frutas e de gomas, pirulito de frutas, algodão-doce, geleia de frutas, goiabada, etc) e derivados da mandioca. Em relação às bebidas, sucos de frutas artificiais, refrigerantes isentos de aspartame, groselha, café, chá, etc. são exemplos do que é permitido.
Por outro lado, os alimentos vedados para essas pessoas são aqueles que possuem nível elevado de fenilalanina. São exemplos, carne e derivados, feijão, ervilha, soja, grão-de-bico, lentilha, amendoim, leite (e derivados),alimentos que possuem aspartame na sua composição, etc.
Ainda existem alimentos que possuem um nível intermediário de fenilalanina (10 - 200mg PHE/100g do alimento), porém não estão totalmente vedados da dieta dos fenilcetonúricos, mas devem ser consumidos cuidando-se para que não ultrapassem os níveis permitidos da FEL no sangue. Fatores como a idade e tolerância particular também devem ser observados. São exemplos de alimentos que se enquadram nessa categoria: massas feitas sem ovos e com farinha de trigo de baixo teor de proteína, arroz, batata-inglesa, batata-doce, beterraba, brócolis, cenoura, frutas, etc.
Algumas das possíveis razões pelas quais os fenilcetonúricos não aderem ao tratamento são a falta de incentivo para seguir a dieta já que a mesma possui custo elevado e necessita de suplementação, a pressão social que gera exclusão desses portadores, o restrito índice de alimentos nutritivos que possuem um menor teor de tirosina. Por outro lado, quando aderem adequadamente a dieta sugerida, são pessoas capazes de manter relações sociais normais, sem desvios comportamentais, aptos a atividades laborais e recreativas como qualquer outro indivíduo.
Fenilcetonúria e a implicação nas relações sociais
Ser um portador de fenilcetonúria é praticamente levar para toda a vida a responsabilidade com os cuidados necessários que a doença exige como a alimentação restrita e a necessidade da obrigação de segui-la para evitar danos à própria saúde e, além disso, ter a consciência de que ainda não dispomos de um suporte em todas as esferas (municipal, estadual, nacional e até mesmo global) para enfrentar as barreiras impostas pela doença, como a falta de informação nos rótulos dos alimentos, má distribuição de centros de atendimentos e a falta de conhecimento da população em geral que muitas vezes não sabe como agir com esse portador.
Por outro lado, os pais que possuem filhos fenilcetonúricos também passam por diferentes etapas de aceitação até chegar numa relação benéfica para ambos, de apoio e ajuda, consistindo numa superação de fatores conflitantes: inicialmente, a maneira pela qual a notícia de um filho portador da doença é recebida pelos pais – os quais sonhavam em ter um filho saudável – e, posteriormente, encarando o novo, já que o filho necessita de cuidados e depende muito dos pais e, por fim, adequando-se a essa nova vida.
Referências:
1.BRASIL. Ministério da saúde – Secretaria de atenção à saúde. Portaria SAS/MS nº 712, de 17 de dezembro de 2010. Disponível em: <http://portal.saude.gov.br/portal/arquivos/pdf/pcdt_fenilcetonuria.pdf>. Acesso em: 14 nov. 2012.
2.CECIL. Desordens no Metabolismo da fenilalanina e da tirosina. Tratado de Medicina Interna. 22. ed., Edit. Elsevier. São Paulo, 2005. Cap 218, p. 1487-1488.
3.DE MIRA Nádia; MARQUEZ Ursula. Importância do diagnóstico e tratamento da fenilcetonúria. Revista Saúde Pública, v. 34 (1): pag. 86-96, São Paulo 2000. Disponível em: <http://www.scielo.br/pdf/rsp/v34n1/1387.pdf>. Acesso em: 14 nov. 2012.
4.MONTEIRO, Lenice; CÂNDIDO, Lys. Fenilcetonúria no Brasil: evolução e casos. Revista de Nutrição, v. 19 (3), Campinas, Mai. e Jun. 2006. Disponível em: <http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1415-52732006000300009>. Acesso em: 14 nov. 2012.
5.SOUZA, Kátia et al. O conhecimento das mães a respeito da fenilcetonuria e do teste do Pezinho. Moreira Jr editora. Disponível em: <http://www.moreirajr.com.br/revistas.asp?id_materia=2079&fase=imprime>. Acesso em: 14 nov. 2012.
Links relacionados:
Anuário da produção acadêmica docente - Enfrentamento dos pais no acompanhamento dos filhos portadores de fenilcetonuria.http://www.sare.anhanguera.com/index.php/anudo/article/view/630/709
Minha vida – Fenilcetonúria: causas, sintomas e tratamento. http://www.minhavida.com.br/saude/temas/fenilcetonuria
Portal ANVISA – Esclarecimentos sobre a fenilcetonúria. http://portal.anvisa.gov.br/wps/wcm/connect/d096c4804ad89a85a95aafa337abae9d/Esclarecimentos_sobre_a_fenilcetonuria.pdf?MOD=AJPERES
Revista UFGV - Teste do Pezinho: por que coletar na alta hospitalar? http://www.revistas.ufg.br/index.php/fen/article/view/781/875
Texto sobre fenilcetonúria. http://www.uff.br/disicamep/fenil.htm
Vídeo - Fenilcetonúria, tire suas dúvidas. http://www.youtube.com/watch?v=l48nvf3dLQE